Hemoglobinopathies
The hemoglobinopathies are any of a group of diseases characterised by abnormalities, both quantitative and qualitative, in the synthesis of hemoglobin (Hb). Most of them are genetically inherited but occasionally they can be caused by a spontaneous mutation. They are the world's most common mono-genic, autosomal, and recessive disease in man.
For a hemoglobinopathy disease condition to exist, an abnormal hemoglobin or thalassemia must be inherited from both parents resulting in a homozygous or double (compound) heterozygous condition.
A trait condition (carrier state) exists when a person inherits one normal Hb gene and one abnormal Hb gene. This person is healthy under normal circumstances and often is not aware they are carrying abnormal hemoglobin (there are some rare exceptions and extenuating circumstances where trait carriers can have symptoms).
Subdivisions of hemoglobinopathies - Structural hemoglobin variants:
- Quantitative defects caused by a reduced/imbalanced synthesis of a normal globin chain are referred to as the “Thalassemias”.
- Qualitative defects caused by the normal synthesis of an abnormal globin chain, often due to single amino acid substitutions in either the alpha or beta globin chains. A common example is the Glu to Val mutation at position six of the beta globin chain in sickle cell disease
- Hereditary persistence of fetal hemoglobin:
- Genetic defects in the switch from fetal to adult hemoglobin synthesis
- Fetal hemoglobin persists into adult life
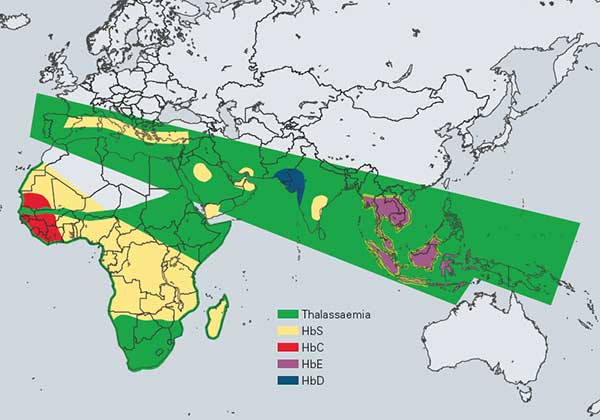
Thalassemias
The term thalassemia is used to describe globin gene disorders that result from a diminished rate of synthesis of one or more globin chains and consequently a reduced rate of synthesis of the hemoglobin or hemoglobins of which that chain constitutes a part; α thalassemia indicates a reduced rate of synthesis of the α globin chain; similarly, β, δ, δb and εγδβ thalassemias indicate a reduced rate of synthesis of the β, δ, δ+β and ε+γ+δ+β chains, respectively. Thalassemia is the most common single gene disorder known.
β-Thalassemia The β-thalassemias are a group of conditions resulting from a reduced rate of synthesis of the β globin chain. More than 200 b gene mutations have been identified, occurring in a wide range of ethnic groups, and within each geographic population there are unique mutations.
β thalassemia mutations are divided into two broad categories, β0 (β-zero) thalassemia and β+ (β-plus) thalassemia. In β0 thalassemia there is either an abnormal gene that is not expressed or, less often, gene deletion. In β+ thalassemia there is reduced, but not absent, expression of the abnormal gene so that even in the homozygous state there is still some hemoglobin A production.
β-thalassemia can be divided into three general categories: - β-thalassemia trait: This state is characterised by heterozygosity of one deleted or mutated gene and one normal functioning gene, and may therefore also be referred to as β thalassemia minor. Individuals with this trait are usually completely asymptomatic.
- β-thalassemia intermedia: This state refers to a clinical phenotype with diverse genetic explanations. These individuals will have a homozygous or heterozygous β globin mutation that causes a decrease in β chain production, but not to the degree that chronic transfusion therapy is necessary. In comparison with a typical patient with β-thalassemia trait, there are significant clinical problems.
- β-thalassemia major or Cooley’s anemia: β-thalassemia major refers to patients with homozygosity or compound heterozygosity for β-thalassemia who are dependent on blood transfusions to maintain life beyond early childhood.
Diagnosing thalassemia traits and diseases Thalassemia may be suspected if an individual shows signs that are suggestive of the disease. However, in all cases, laboratory diagnosis is essential to confirm the exact diagnosis and to allow for the provision of exact genetic counselling about recurrence risks and testing options for parents and affected individuals. Likewise, screening is recommended to determine trait status for individuals of high risk ethnic groups.
The following tests are used to screen and diagnose thalassemia disease and/or trait:
References: - Adrain Stephens; Haemoglobinopathies; The Biomedical Scientist; 2004, July, 1-4
- Anita J. Catlin; Thalassemia: The facts and the controversies; Pediatric Nursing, November-December 2003, 29 (6), 447-451
- Barbara J. Bain; Haemoglobinopathy Diagnosis; 2001 Blackwell Science Ltd.
- Nancy F. Olivieri; The β-thalassemias; The New England Journal of Medicine, 1999, July 8, 341 (2), 99-109
- P.C.Giordano, M.Herruer, W.Huisman, J.G.J.Pouwels, J.Smit, N.Verhoef, P.Wijermans; Rapport van de werkgroep hemoglobinopathieën van de vereniging hematologische laboratoriumonderzoek. Enquêteresultaten en aanbevelingen t.b.v. dragerschapdiagnostiek en preventie van de hemobglobinopathieën. Uitgegeven door de leden van het landelijk werkgroep hemoglobinopathieën van de vereniging van hematologische laboratoria.
A lot of information was obtained from the following Internet websites: